Current Challenges in Antifungal world: Morbidity and mortality from invasive fungal infections are significant, and largely caused by two genera of fungal pathogens: Candida and Aspergillus. Candida species are the 4th most common pathogen isolated in all bloodstream infections. Treatment for invasive candidiasis has a limited (50-70%) success rate, and this is typically only in the healthiest patients. The incidence of invasive aspergillosis due to A. fumigatus has increased three-fold in the last decade and its mortality has risen by over 300%. Moreover, current therapy for invasive aspergillosis has a lower 40-50% treatment success rate. Invasive aspergillosis is consistently a leading killer in immunocompromised patients, and invasive mold infections (fusariosis, scedosporosis, and mucromycosis) have even higher mortality rates and no effective therapeutic options.
Our Motto: Amphotericin B (AmB) is an exceptionally promising starting point, because this drug has potent and dose-dependent fungicidal activity against a broad range of fungal pathogens and has evaded resistance for over half a century. The problem is that AmB is exceptionally toxic, which limits its use to low-dose protocols that often fail to eradicate disease. An AmB derivative that retains potent, broad spectrum, and resistance-evasive fungicidal activity but lacks dose-limiting toxicities would enable a new high-dose treatment paradigm with improved clinical efficacy.
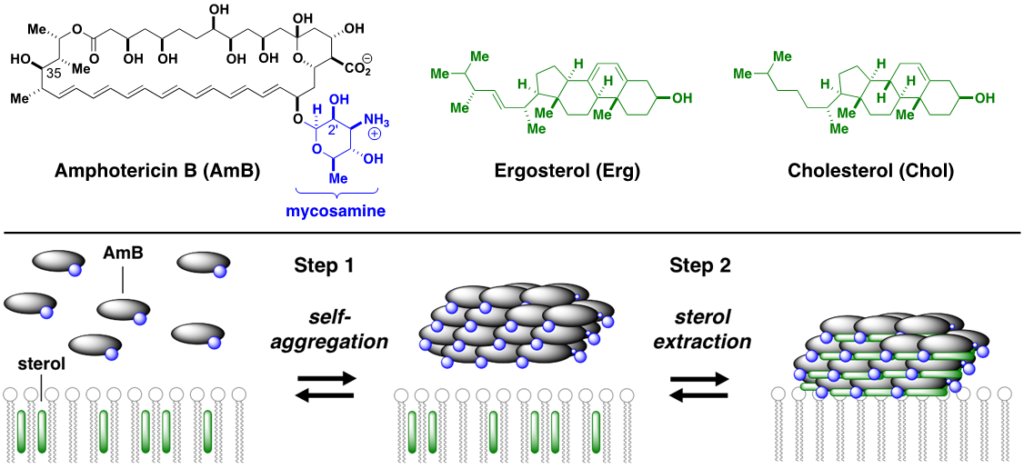
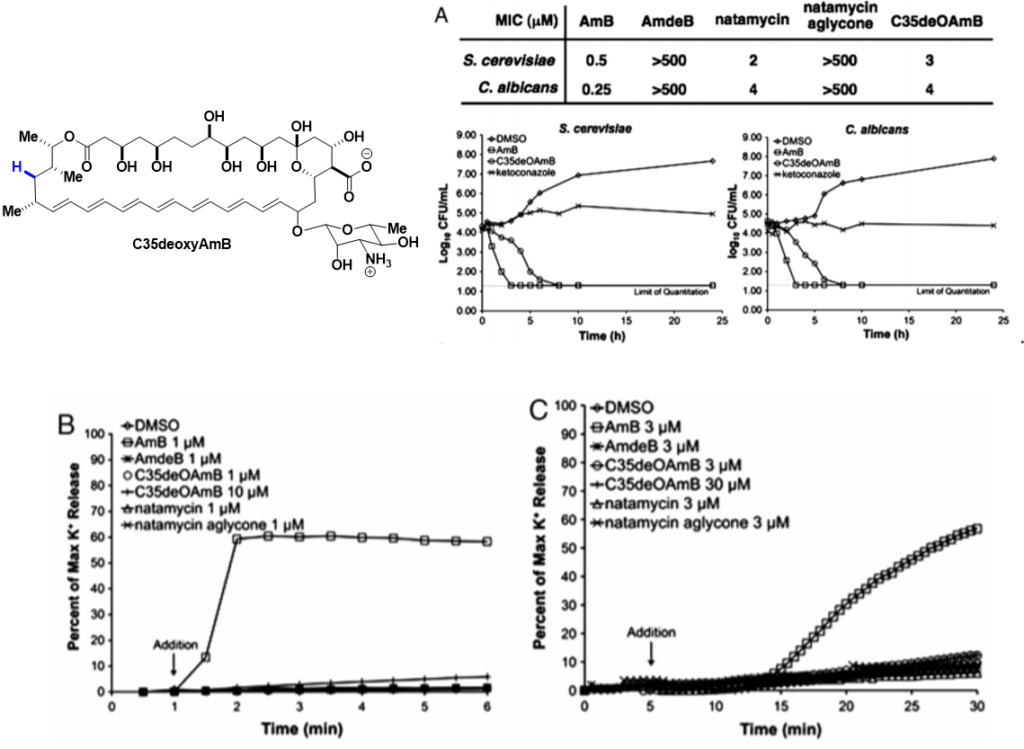
What we do: More than half a century of unsuccessful attempts to develop a clinically viable less toxic derivative of AmB were fundamentally misguided by the widely accepted mechanistic model which asserts that this natural product primarily kills cells by permeabilizing membranes. Enabled by our development of frontier synthesis methods for efficient modification of complex natural products., we alternatively discovered that AmB primarily kills fungal and human cells by binding ergosterol and cholesterol, respectively; channel formation is not required. By selective deletion of C35-OH group of AmB, we have shown that the new molecule (C35deoxyAmB) does not show ion channel formation yet retains its antifungal property which is consistent with a “sterol sponge” model, whereby AmB self-assembles into a large extramembraneous aggregate and rapidly extracts physiologically vital sterols from fungal and human cells, thereby causing cell death. Frontier SSNMR studies (w/ Chad Rienstra at UIUC) further revealed key insights into the structure of AmB sponge-sterol complexes.
Figure 1. Top, Chemical structure of amphotericin B, the primary fungal sterol-ergosterol, and the primary human sterol-cholesterol. Middle, Two-step ”sterol-sponge” model for the cytocidal action of AmB. Bottom, Chemical structure of C35deoxyAmB and its antifungal and ion channel forming properties.
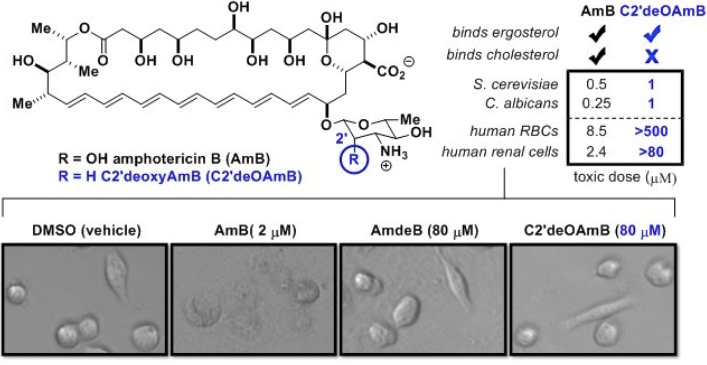
Our Key Findings: To probe its predicted role in sterol binding, we synthetically deleted the hydroxyl group at the C2’ position on the mycosamine appendage. The resulting derivative, C2’deOAmB, was found to bind ergosterol but, within the detection limits of isothermal titration calorimetry (ITC), not cholesterol. Consistent with the sterol sponge model, this derivative retained good activity against yeast but, most importantly, was non-toxic to human red blood cells and primary hRECs.
Figure 2. C2’deOAmB shows potent activity to yeast but non-toxic to human cells
To rationalize the selective toxicity of C2’deOAmB for fungal vs. human cells, we proposed a model in which the C2’-OH stabilizes a conformer of AmB that readily binds both ergosterol and cholesterol. We hypothesized that deletion of this hydroxyl group favors a shift to a different conformer or set of conformers which retain the capacity to bind ergosterol but not the more sterically bulky cholesterol. Alternatively stated, this model suggests that deletion of the C2’OH of AmB causes a small molecule-based allosteric effect that results in ligand-selective binding.
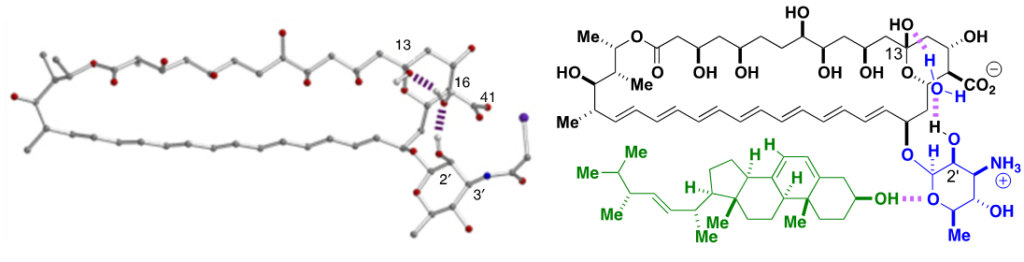
Figure 3. Left, X-ray crystal structure of N-iodoacyl AmB. Right, Proposed structural model for AmB-Erg complex. A similar model is proposed for Chol.
C2′deOAmB thus revealed that a modification of AmB could lead to binding of ergosterol but not cholesterol, demonstrated that such selective binding translates to a dramatically improved therapeutic index in vitro, and suggested a potentially predictable structural model for how such selectivity was achieved. However, several key questions remained unanswered. Foremost, it was unclear if these in vitro results would translate to an improved therapeutic index in vivo. Moreover, it was unclear if an improved therapeutic index would come at the cost of a decreased capacity to evade resistance. As described above, one of the most remarkable features of AmB is that despite half a century of utilization, there has been no significant emergence of pathogen resistance in the clinic. It has long been suspected that the exceptional toxicity of AmB and its capacity to evade resistance were linked. In fact, less selective drugs are generally associated with increased capacity to evade resistance, but also with increased toxicity. Thus, it was unclear if a less toxic AmB derivative would be more vulnerable to pathogen resistance. Answering both of these questions required access to larger quantities of a non-toxic AmB to support a wide range of in vitro and in vivo studies. Thus, in order for an AmB derivative to have a chance to be advanced for clinical development, it must be accessible in an exceptionally efficient manner, and the lack of scalable access to C2′deOAmB limited its further development. We thus sought a different AmB derivative that would demonstrate a similar improvement in therapeutic index but would be potentially accessible on the metric-ton scale.
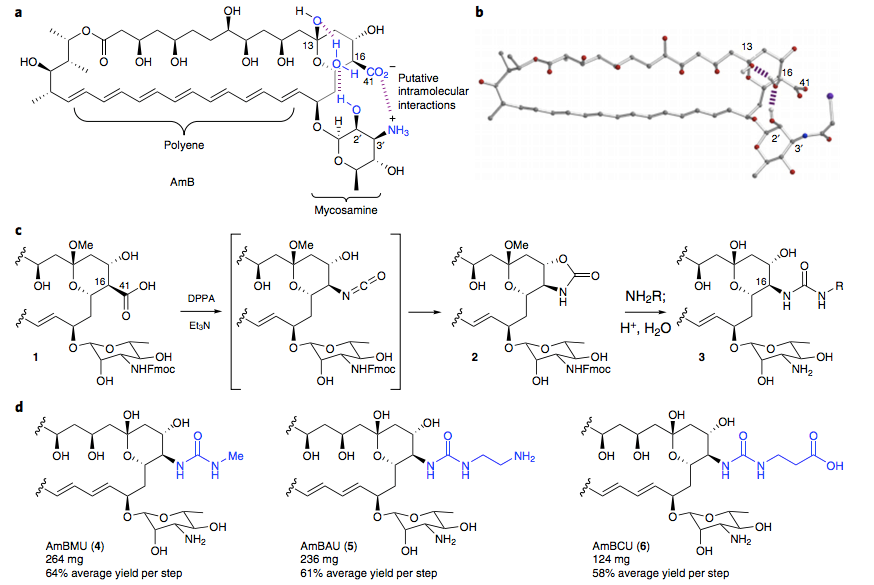
The ligand-selective allosteric model and the aforementioned crystal structure suggested a potential path forward. Specifically, in addition to the water-bridged hydrogen bonding interaction between the C2′ hydroxy and C13 hemiketal groups, the X-ray crystal structure of N-iodoacetyl AmB suggested to us that an intramolecular salt bridge between the C41 carboxylate and what would be a C3′ ammonium ion may also help stabilize the ground-state conformation of AmB predicted to bind both ergosterol and cholesterol. We reasoned that perturbing this putative interaction may represent an alternative way to cause a similar shift in the position of the sugar relative to the polyene macrolide core that would lead to the same selective sterol binding that had been achieved with C2′deOAmB. Importantly, many different types of modifications to alter this putative intramolecular salt bridge could be accessible in just a few steps from the already ton-scale fermented natural product.
Due to its ease of modification, the C41 carboxylate has been a common site for derivatization, and has previously yielded derivatives of AmB with encouraging but modest improvements in its therapeutic index. However, we noticed that all of these derivatives had retained the C16–C41 carbon–carbon bond. As part of contemporaneous studies, we discovered that diphenyl phosphoryl azide promotes an efficient Curtius rearrangement on a minimally protected AmB, cleaving the C16–C41 bond to form an isocyanate intermediate. The isocyanate intermediate is immediately trapped by the neighboring C15 hydroxy forming an isolable oxazolidinone intermediate. Surprisingly, this oxazolidinone is quite easily opened under mild conditions with amines leading to a new class of AmB derivatives, the AmB ureas, which can be accessed in just three steps from the natural product. Guided by the ligand-selective allosteric effects model described above, we questioned whether, like C2′deOAmB, these AmB ureas might selectively bind ergosterol over cholesterol.
Figure 4. Design and synthesis of AmB ureas.
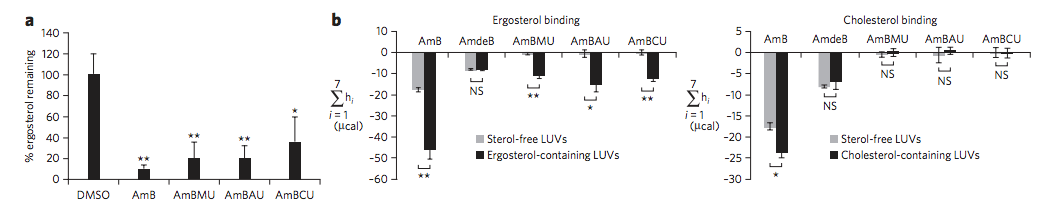
We tested the sterol binding of the urea derivatives via ITC and found that they retain the capacity to bind ergosterol. However, within the limits of detection, we observed no binding to cholesterol. We then evaluated these urea derivatives in antifungal and hemolysis assays and compared them to many previously reported modifications of the C41 carboxylate, including methyl esterification (AmBME), reduction to a methyl group (MeAmB), and conversion into a methyl amide (AmBMA). We also resynthesized a doubly modified derivative (AmBTABA) that represented one of the largest previously reported improvements in in vitro therapeutic index. These derivatives all demonstrated modest improvements in therapeutic index compared to AmB. However, in stark contrast, the AmB ureas showed a remarkable decrease in toxicity to human red blood cells with both the methyl urea (AmBMU) and aminoethyl urea (AmBAU) failing to show complete hemolysis even up to 500 μM. Interestingly, the only structural difference between AmBMU and AmBMA is a protonated nitrogen atom between the C16 and C41 carbons.
Figure 5. Erg extraction and ITC experiments to Erg and cholesterol of AmB ureas.
We further probed the activity of these AmB ureas against a panel of pathogenic Candida, Cryptococcus, and Aspergillus strains, including strains that are resistant to the azole class of antifungals. In all of the strains, the AmB ureas maintained potent antifungal activity. Final in vitro testing looked at the renal toxicity of the urea compounds against primary RPTECs and an immortalized human renal cell line (hTERT1 RPTECs). In both types of renal cells, we saw diminished toxicity compared to AmB. This promising series of in vitro assays led us to seek out a collaboration with David Andes at the University of Wisconsin at Madison, who has developed the most widely employed mouse model of invasive candidiasis.
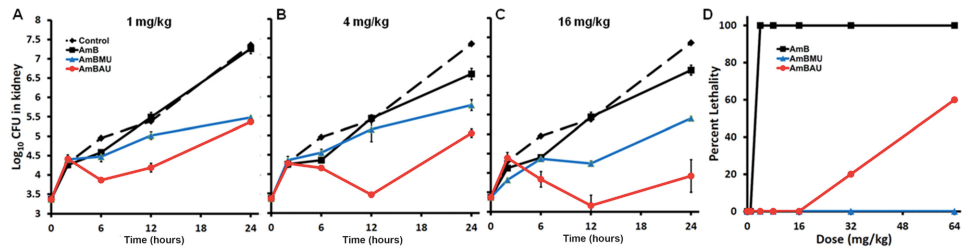
Figure 6. Time course of fungal burden in the kidneys of neutropenic mice infected with C. albicans following a single intraperitoneal injection of AmB, AmBMU, or AmBAU at dosages of (A) 1 mg per kg body weight, (B) 4 mg per kg body weight, or (C) 16 mg per kg body weight. (D) Dose-response lethality following a single intravenous injection of AmB, AmBMU, or AmBAU at dosages ranging from 0.5 mg per kg body weight to 64 mg per kg body weight.
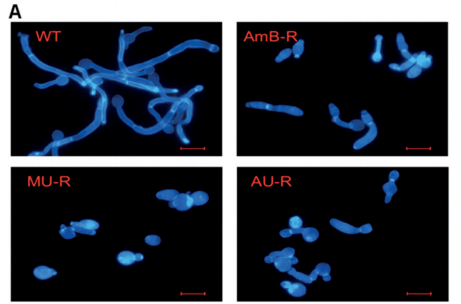
Neutropenic mice infected with a clinical isolate of C. albicans were treated with AmB, AmBMU, or AmBAU at 1, 4, or 16 mg per kg. We observed a pronounced reduction in fungal burden by both AmBMU and AmBAU compared to AmB, especially at the 16 mg per kg dosage at 24 hours post-treatment. To study in vivo toxicity, we monitored lethality from intravenous injections of AmB or the urea derivatives to healthy and uninfected mice. At just 4 mg per kg, AmB killed all of the mice within seconds. It was not until 64 mg per kg administration that lethality exceeded 50% from AmBAU, and even at 64 mg per kg, no lethality was observed for AmBMU. Finally, we teamed up with Susan Lindquist and her colleagues, Ben Vincent and Luke Whitesell, at the Massachusetts Institute of Technology to determine whether the improved therapeutic index observed for these AmB ureas came at the cost of increased vulnerability to resistance. AmBMU and AmBAU were tested against a panel of labgenerated C. albicans strains with mutations to various ergosterol biosynthesis genes.40 Similar to AmB, only the erg2, erg6, and erg3erg11 mutants were resistant to the urea derivatives and these mutants are known to be avirulent. We sought out other potential mutations that may incur resistance by gradual resistance-selection with AmB and both urea derivatives. We then sequenced the full genome of each of the rare resistant mutants that we were able to generate against AmB, AmBMU, and AmBAU. For all three compounds, resistance mutations were primarily found in the ERG2 or ERG6 locus, and no unique resistance mutations were observed with AmBMU or AmBAU relative to AmB.
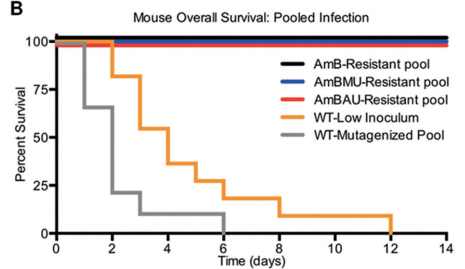
Figure 7. (A) Filamentation response for wild-type, AmB-resistant (AmB-R), AmBMU-resistant (MU-R), and AmBAU-resistant (AU-R) C. albicans. (B) Mice survival following injection of AmB-resistant strains, AmBMU-resistant strains, AmBAU-resistant strains, parental wild-type (WT-low) strains, or mutagenized wild-type mutants (WT-mutagenized) strains.
Our Next Direction:In our next generation approach, interlinking the toxicity evading derivatization and potency promoting functionalization demonstrated unprecedented improvement in therapeutic index. Further preclinical studies on the lead compounds are currently ongoing.